Wan Lab Research Focus
Single Cell Analysis
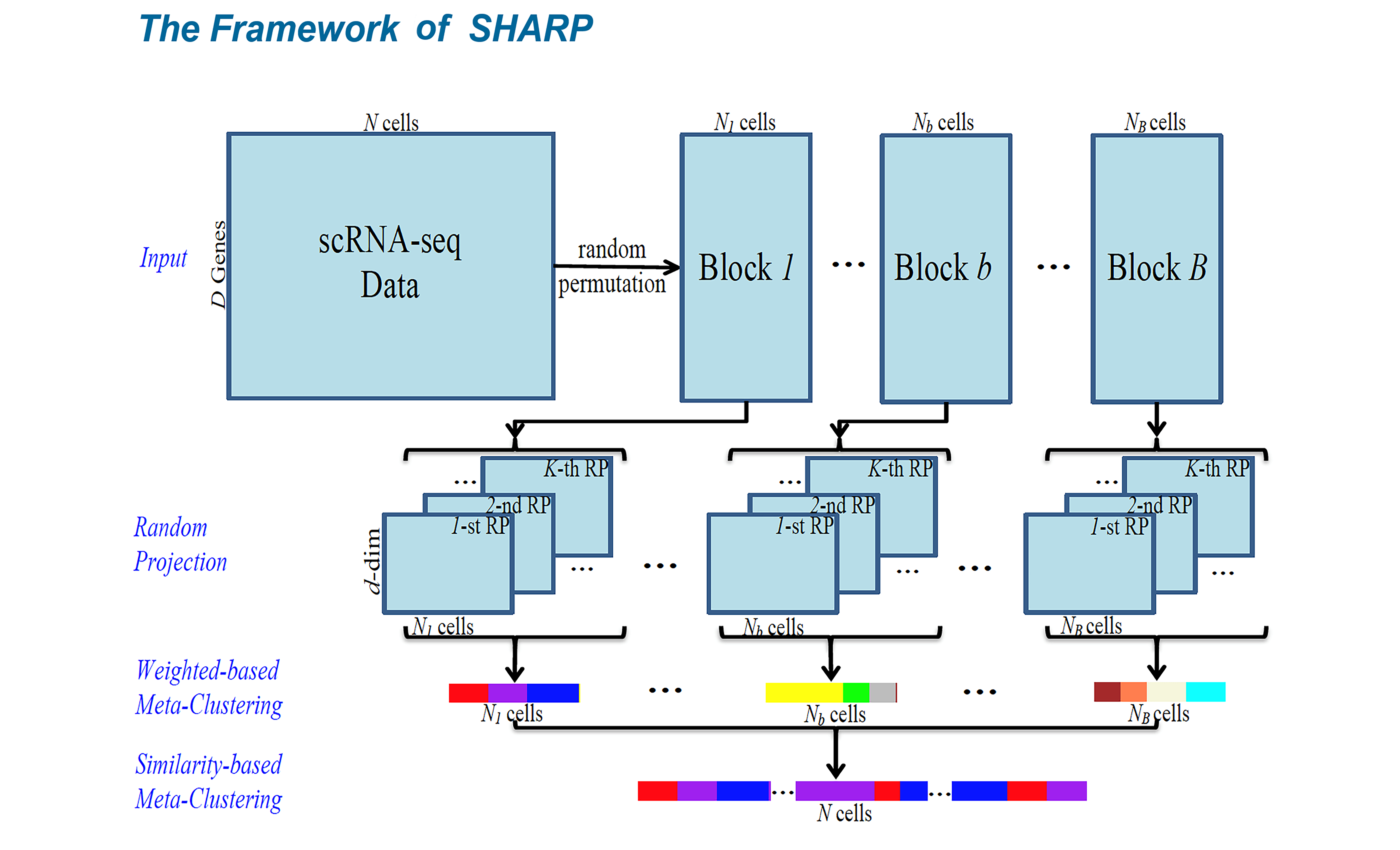
As one of the most essential and far-reaching technologies in recent decades, single-cell sequencing has been selected as “Method of the Year” by Nature Methods three times, namely single-cell sequencing for 2013, single-cell multimodal omics for 2019 and spatial transcriptomics for 2020. By enabling profiling at the individual-cell level, single-cell sequencing enables researchers to characterize novel cell types and interrogate intra-population heterogeneity. Given the rich information single cell data reveal, there are many challenges related to single cell analysis that remain to be addressed, including but not limited to, clustering, batch effect correction, multi-modal data integration, trajectory inference, RNA velocity, etc. Our laboratory has made some contributions to the topic of clustering, where I proposed the first computational model (i.e., SHARP) that is capable of processing 10-million cells for single-cell data analysis fast and accurately. The remaining challenges are a major focus in our lab.
Multi-Omics Analysis
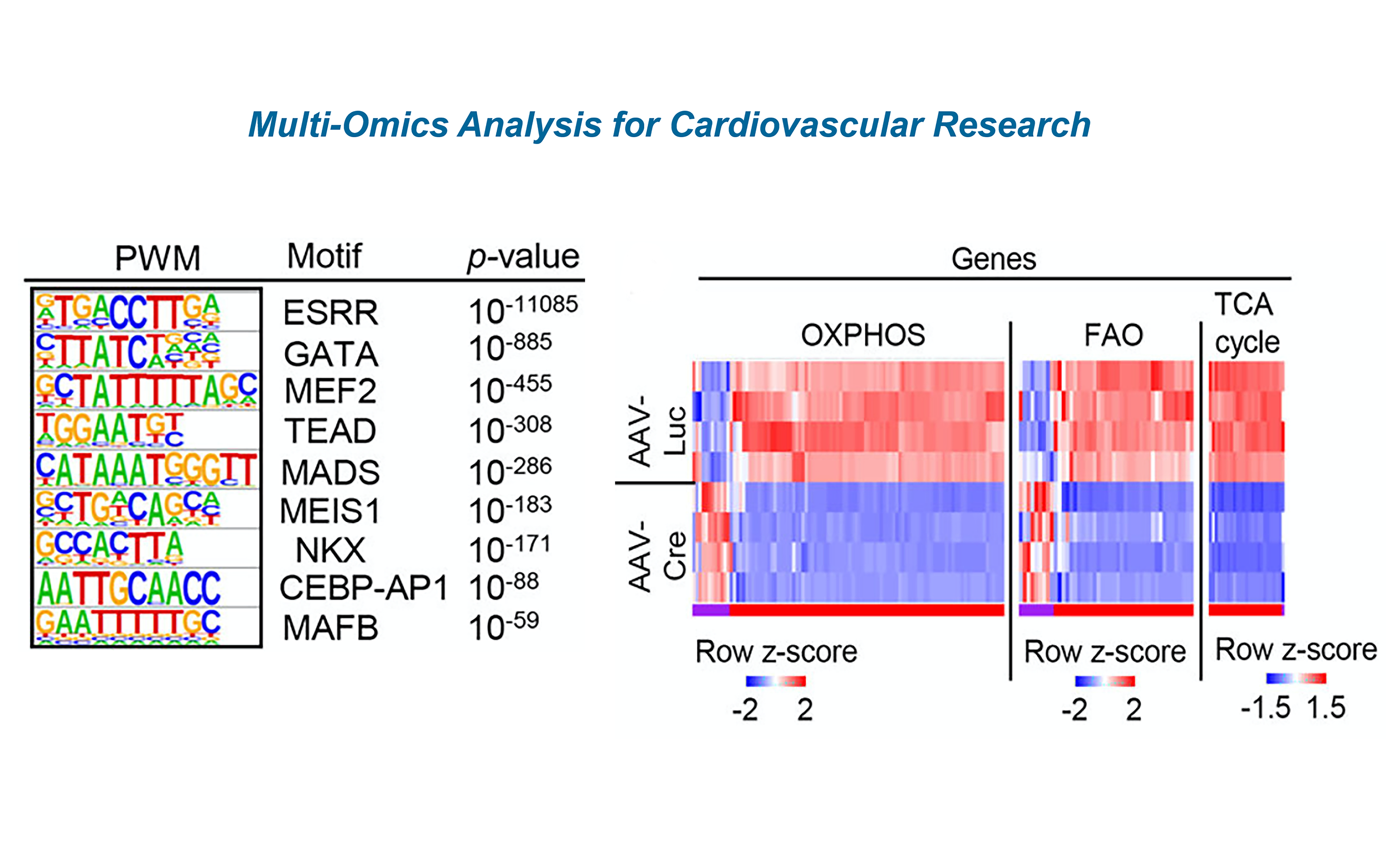
The integration of multi-omics including genomics, transcriptomics, epigenetics, proteomics, metabolomics and interactomics in both bulk and single cell data, can contribute to pinpointing biomarkers of disease and physiology, and to deciphering mechanisms of associations among genotypes, phenotypes and envirotypes. Commonly, conventional models first analyze each type of omics data independently and then leverage potential (yet perhaps weak) connections among them. These models heavily rely manual interventions and different persons might yield different interpretations. To overcome these problems, our laboratory will develop computational models to automatically learn heterogeneous multi-omics data. For example, some potential machine learning algorithms include multi-kernel learning, co-learning, multimodal representation, and joint representation. The first two categories of models simultaneously learn the informative features from the multi- omics data, whereas the latter two categories automate the representations of multi-omics data in the same feature space. For single-cell multi-omics data, we can also combine our existing machine learning models to deal with high-dimensionality and big-data problems.
Spatial Transcriptomics 
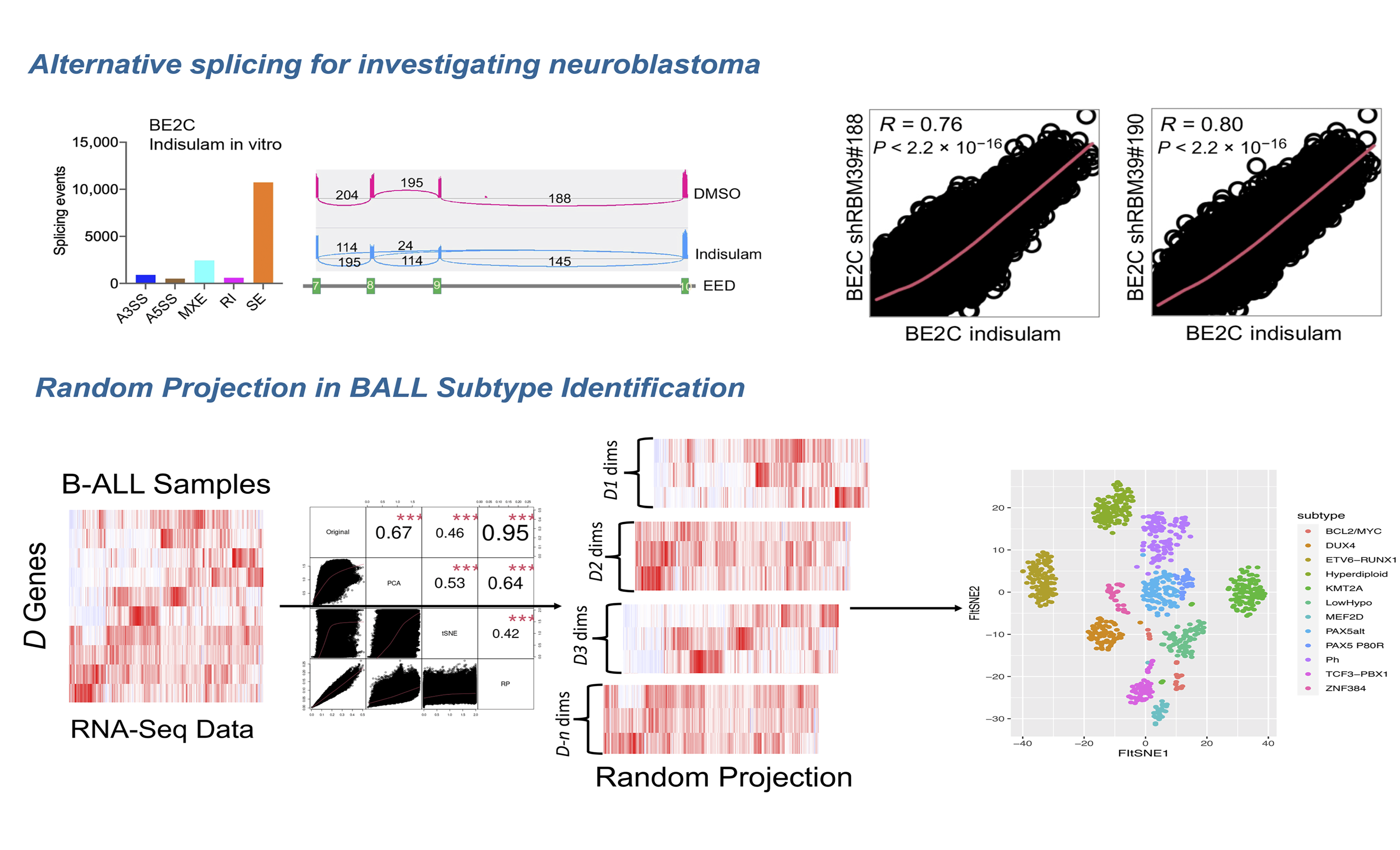
In spatial transcriptomics, elucidating single-cell heterogeneity while also retaining the spatial information is crucial for understanding key aspects of cell development and differentiation, cell- cell interaction, tumorigenesis, and cancer progression, etc. Spatial transcriptomics has also been used to determine subcellular localization of mRNA molecules, which is highly related to my previous research on protein subcellular localization. More importantly, with the coupling of spatial transcriptomics and single cell sequencing, we anticipate multimodal spatial profiling of transcriptome, genome, and proteome simultaneously. Given that both sequencing and imaging data were generated in spatial transcriptomics, we believe computational models can play significant roles in spatial transcriptomics. Our laboratory leverages machine learning and bioinformatics methods to tackle these problems.
Machine Learning for Cancer Research, Intelligent Healthcare, and Precision Medicine
With the explosion of heterogeneous and big data in biology and medicine, machine learning has become an essential tool for cancer research, disease diagnostics, therapeutic development, and drug discovery. In addition to the omics data, our laboratory is also interested in tackling other basic, translational and clinical data in various data formats (e.g., sequencing, electronic health records, images, etc) by artificial intelligence, machine learning and data science methods to facilitate intelligent healthcare and precision medicine. By establishing extensive collaborations with clinicians, doctors, radiologists and pathologists, we develop and/or leverage computational models to help unravel the mechanisms of pathogenesis, tumorigenesis, disease diagnosis and prognosis, treatment design and drug discovery.
Bioinformatics Tool Development
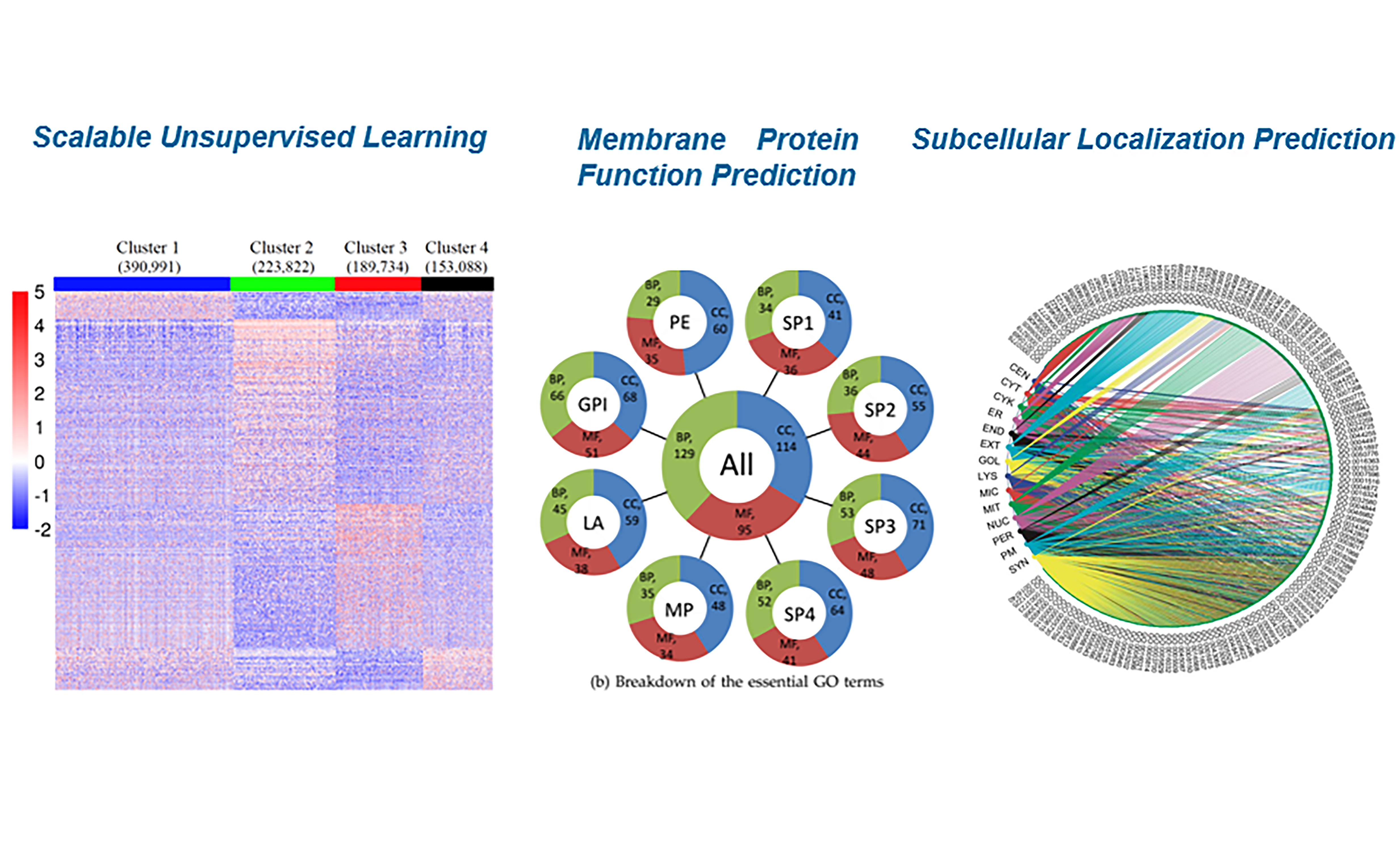
Bioinformatics tool development is one of the thematic research directions in Wan Lab. Being equipped with both machine learning and bioinformatics, Wan Lab is dedicated to develop bioinformatics tools in various domains ranging from basic science, translational research, to clinical applications, including but not limited to single cell analysis, antimicrobial peptide identification, cancer subtype prediction, cancer diagnosis and prognosis, protein subcellular localization, membrane protein function prediction, etc.
Ongoing Research Projects
Projects in Wan Lab focus on develop artificial intelligence/machine learning and bioinformatics/computational biology based methods for single cell analysis, multi-omics analysis, spatial transcriptomics, cancer research, intelligent healthcare, and precision medicine.
- Single Cell Analysis
- Ultra-large-scale clustering for single cell transcriptomics data analysis
- Single Cell Multi-Omics Integration
- Batch effect correction
- Rare cell type detection for large-scale single cell data
- Leukemia Subtype Identification (ALL, AML, etc)
- Cancer Disparities by AI/ML Methods
- Antimicrobial Peptide (AMP) Identification
- Neuroimaging Biomarker Identification for Alcohol Use Disorder (AUD)
- Integrating Multi-Omics and Imaging Data for Neurological Disorders (Alzheimer’s Disease, Parkinson Disease, etc)
- Prostate Cancer Research
- Lung Cancer Characterization
- Proteomics Data Analysis
… and more.